
F PLUS HEALTHCARE TECHNOLOGIES | INSIGHTS FOR PHARMA MANUFACTURERS
A recent industry recall traced a sterile injectable contamination event back to a single weld bead – a small section of internal pipe surface that had never been polished. The machine was GMP-compliant. It had passed every inspection. It had cleared validation.
This kind of failure pattern is more common than the industry likes to admit. Most pharmaceutical contamination events don’t come from broken protocols or careless operators. They come from engineering decisions made by machinery manufacturers years earlier, and never questioned.
The conversation around contamination usually circles cleanrooms, SOPs, and operator hygiene. All of it matters. But the contamination story almost always begins much earlier. It begins in the design and engineering of the machine itself.
This is what most pharmaceutical equipment manufacturers get wrong, and what it actually takes to engineer for zero contamination.
Compliance Is a Floor, Not a Ceiling
Most pharmaceutical machinery manufacturers design to pass inspection. Passing inspection is not the same as engineering for zero contamination.
Compliance defines the minimum acceptable standard. A GMP-compliant machine can, and often does carry subtle engineering flaws that allow contamination to survive validated cleaning cycles. The gap between “compliant” and “contamination-controlled” is where recalls live.
That gap is built from five engineering decisions most machinery manufacturers underweight.
1. Dead Legs in Piping Systems
A dead leg is any section of pipe where fluid stops circulating. Bacteria settle. Biofilm forms. Cleaning cycles can’t reliably reach it.
WHO TRS 929 sets the rule clearly: dead legs in WFI, product transfer lines, and CIP loops should not exceed three pipe diameters (3D). The standard exists because the science is settled.
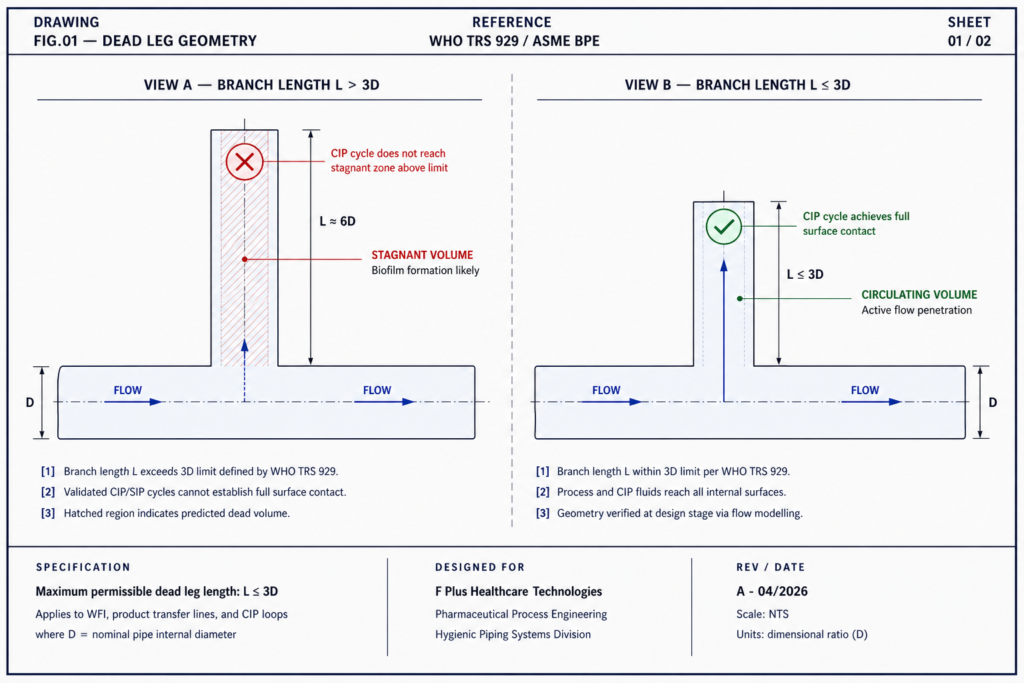
Figure 1 – Non-compliant vs compliant dead-leg geometry. Branch length above 3D creates stagnant zones.
Yet dead legs get introduced routinely, most often during facility expansions, when a new line is tied into an existing system without a full engineering review. The expansion passes drawings. It doesn’t pass real flow analysis. Six months later, a deviation investigation finds the source.
Designing a system without dead legs is harder. It requires pre-built routing models, sloped piping, and zero-static-pocket geometry from the first drawing. It is also non-negotiable.
2. Surface Finish and Material Integrity
Electropolished 316L stainless steel with Ra ≤ 0.6 µm is not an aesthetic specification. At that finish, microscopic crevices where bacteria anchor and biofilm forms are eliminated. CIP and SIP cycles can finally do what they were validated to do.
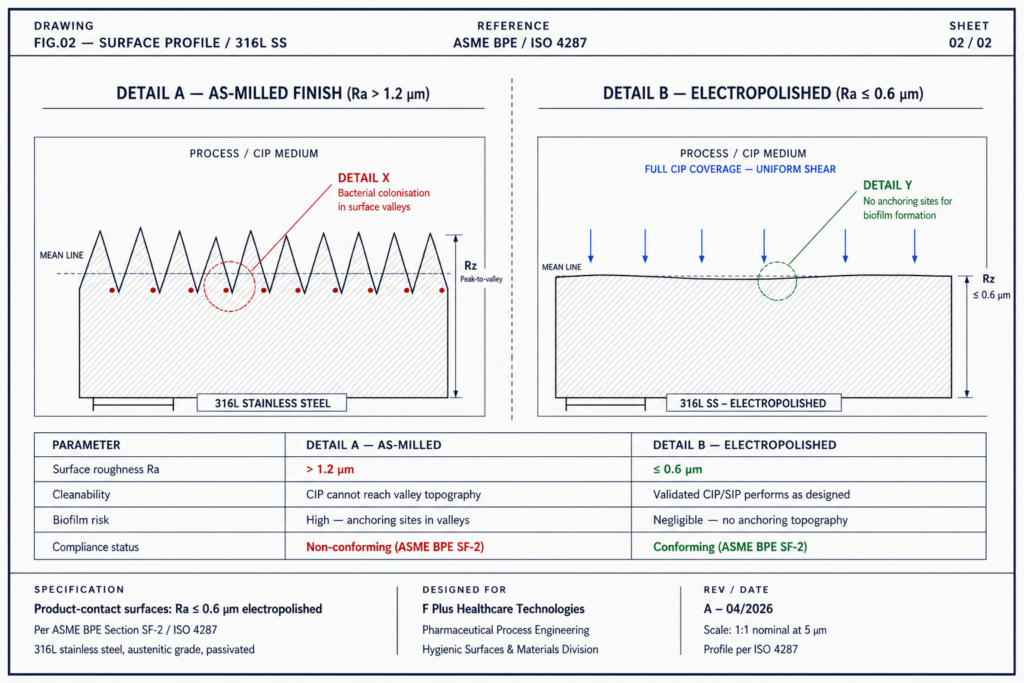
Figure 2 – Cross-sectional view: rough mill finish vs electropolished surface at the same magnification.
Procurement teams routinely substitute lower-grade finishes to cut cost. The savings are visible in the PO. The cost shows up later, in failed swab tests, in extended cleaning cycles, in batches that never ship.
Material chemistry matters just as much as finish. 316L is the industry standard, but the metallurgical condition of that steel: heat treatment, weld quality, passivation determines whether it actually performs as 316L. Internal weld beads that aren’t orbitally welded and post-polished create sheltered zones invisible to spray balls and unreachable by steam.
The same logic extends to elastomers. Gaskets, O-rings, and diaphragm seals are usually specified on pressure and temperature alone. Their extractable and leachable profiles, their compatibility with sanitizing agents, and their long-term swelling behavior under actual process conditions are often evaluated only after installation – when replacement becomes expensive and disruptive.
3. Equipment Geometry and Cleanability
Most pharma equipment is geometry-optimized for process efficiency. Cleanability is treated as a secondary constraint.
The result is predictable horizontal surfaces that pool liquid, blind corners that spray balls can’t reach, bolted assemblies that require disassembly to clean, and tight-tolerance gaps where residue dries between batches. Every one of these is a contamination reservoir hiding inside a validated machine.
This is also where human behavior becomes a contamination factor. When equipment requires excessive manual cleaning, operators take shortcuts. Not because they’re careless, but because they are human. The fastest path is rarely the cleanest one. Equipment that engineers in cleanability removes that trade-off entirely.
ASME BPE guidelines – the global gold standard for hygienic design exist precisely to close this gap. They specify how surfaces, drainage, welds, and assemblies should be engineered for cleanability from the start. Pharma machinery manufacturers who treat ASME BPE as optional are betting against statistics.
4. Internal Airflow and Pressure Dynamics
HEPA filtration, laminar flow, and positive pressure zones get designed carefully at the cleanroom level. Inside individual machines, airflow is often an afterthought.
The consequences are real: pressure differentials between machine compartments that pull contaminated air into sterile zones, filter placement that lets particles bypass critical barriers, and sealed enclosures with poorly modeled air exchange. The cleanroom equipment is class 100. The machine interior, in practice, is not.
Engineering airflow at the machine level requires CFD modeling, validated air-balance studies, and filter placement designed against actual particle behavior, not assumed behavior. Most machinery manufacturers skip this. The few that don’t are the ones whose customers pass aseptic process simulations the first time.
5. Validation Engineered for Worst Case, Not Best Case
CIP and SIP validation protocols are among the most rigorously documented procedures in pharmaceutical manufacturing. They are also among the most commonly misapplied.
CIP validation is typically run on clean equipment. By definition, this proves only that a clean machine can be cleaned. Worst-case soiling studies – running validation against the dirtiest realistic batch condition – are a far better test, and far less common.
SIP validation has a similar blind spot. Steam penetration to all product-contact surfaces is verified during qualification. It is rarely re-verified after equipment modifications, which means a machine that was validated three years ago may no longer match the system it has become.
Add to this the issue of comprehensive traceability. Every metallic component in a contamination-critical system should carry full lifecycle documentation, such as heat numbers, mill certificates, weld logs, and finish reports. Many equipment manufacturers can not produce this on demand. The ones that can are the ones regulators trust.
How F Plus Engineers for Zero Contamination
At F Plus Healthcare Technologies, contamination control is not a quality checkpoint. It is an engineering discipline built into how we design from the first drawing.
Design that’s tailor-made. Standard machines force standard compromises. Our pharmaceutical equipment, across granulation, vessels, water systems, filling lines, and containment is engineered around the customer’s actual process, facility, and product. Dead legs, geometry, and airflow are solved at design stage, not corrected after installation.
Integration across the line. Contamination often hides at handoff points between machines from different vendors. Our Total Process & Packaging approach engineers the line as one system, from raw material to final pack, eliminating the seams where contamination travels.
The Bottom Line
Every contamination event traces back to an engineering decision nobody questioned. A dead leg tolerated. A surface finish was compromised. A CIP cycle validated under ideal rather than worst-case conditions. None of this requires new science. It requires manufacturers willing to engineer better pharmaceutical equipment and customers willing to demand it.
If you are evaluating new equipment, planning a facility expansion, or auditing an existing line, the questions in this blog are the ones worth asking. The right answers separate machinery manufacturers who pass inspection from manufacturers whose products never get recalled.
To see how F Plus can engineer contamination control into your next system, write to Info@fplushealthcare.com. We work with pharmaceutical manufacturers across India, Southeast Asia, the Middle East, Africa, and Europe.
FAQs
1. What causes most contamination events in pharmaceutical manufacturing?
The majority of contamination events are not caused by operator error or broken SOPs – they trace back to engineering decisions made during equipment design. Dead legs in piping, sub-standard surface finishes, poor cleanability geometry, unmodeled internal airflow, and best-case validation protocols are the five recurring root causes. Compliance with GMP guarantees only that a machine meets the minimum standard; it does not guarantee that contamination cannot establish itself.
2. What is a dead leg and why does it matter?
A dead leg is any section of process piping where fluid stagnates instead of circulating freely. WHO TRS 929 specifies that dead legs in WFI, product transfer, and CIP loops should not exceed three pipe diameters (3D). Beyond this, biofilm forms and cleaning cycles cannot reliably penetrate. Dead legs are most often introduced during facility expansions when new lines are tied into existing systems without a full engineering review.
3. Why does electropolished 316L stainless steel matter for hygienic design?
Electropolished 316L with a surface roughness of Ra ≤ 0.6 µm eliminates the microscopic crevices where bacteria anchor and biofilm forms. At rougher finishes, validated CIP and SIP cycles cannot fully reach contamination harbours, regardless of cycle time or chemistry. Surface finish is not cosmetic – it is the foundation that makes hygienic cleaning protocols work.
4. What are ASME BPE guidelines and are they mandatory?
ASME BPE (Bioprocessing Equipment) is the global gold standard for hygienic equipment design. It specifies engineering rules for surface finish, drainage, welds, gaskets, and assemblies in pharmaceutical and biopharma equipment. While not legally mandatory in every jurisdiction, ASME BPE is the de facto standard for any manufacturer building equipment for regulated markets – particularly the US FDA and EU EMA. Treating it as optional is a measurable contamination risk.
5. How should CIP and SIP validation be done correctly?
CIP validation should be performed against worst-case soiling conditions – the dirtiest realistic batch state – not against a clean machine. SIP validation must verify steam penetration to every product-contact surface during qualification, and must be re-verified after any equipment modification. Both protocols should be supported by full lifecycle traceability of metallic components, including heat numbers, mill certificates, weld logs, and finish reports.
Ready to upgrade your contamination control?
Talk to the F Plus Team. Tailor-made. Integrated. Built to scale.


